

NLRP1 & NLRP3: a tale of two inflammasome sensors
Early 2000s, NLRP1 and NLRP3 were the first described inflammasomes, consisting in cytosolic multi-protein complexes that sense danger signals. Activated inflammasomes operate the maturation and release of the pro-inflammatory cytokines IL-1β and IL-18 (1). Although NLRP1 was the first identified member of the NLR (Nucleotide-binding domain and Leucine-rich repeat Receptor) family, its functional relevance remained obscure for many years. Meanwhile, NLRP3 rapidly became the prototypical and best-characterized inflammasome, being linked to a long list of diseases and conditions, including type-2 diabetes, gouty arthritis, and cancer(2). Today, NLRP1 research is reaching new momentum. Viral infections (3,4) and UVB radiation (5) have been identified as natural NLRP1 triggers in humans. Moreover, excessive NLRP1 activation contributes to COVID-19 severity in the context of SARS-CoV-2 infection (4), and NLRP1 gain-of-function mutations result in increased squamous cell carcinoma susceptibility (6). It is tempting to speculate that these new findings might dethrone NLRP3 in the inflammasome realm. Indeed, in some diseases, NLRP3 activation may be a bystander outcome from other inflammasome responses. Deciphering the roles of NLRP1 and NLRP3 in homeostatic and pathological contexts will lead to new therapeutics for microbial and non-infectious diseases. Here, we review the current knowledge about these two sensors and discuss the experimental difficulty in assessing their respective role in pathophysiological contexts.
Of mice and men
NLRP3 and NLRP1 differ in many aspects, including tissue expression, protein structure, and activation mechanisms. Moreover, while NLRP3 is highly conserved between humans and mice, it is not the case for NLRP1.
• Expression
In humans, a single NLRP1 gene is alternatively spliced, isoform 1 being the best studied (7). In contrast, mice exhibit a multi-gene configuration. Most of our current knowledge has focused on the murine Nlrp1b paralog (8). Human NLRP1 is highly expressed in epithelial barriers (i.e. keratinocytes and bronchial epithelial cells), whereas mouse NLRP1B and human/mouse NLRP3 are mainly found in myeloid cells (8).
• Architecture
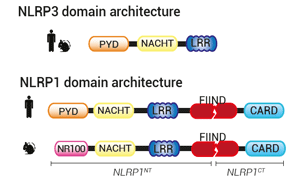
NLR proteins typically feature an N-terminal effector domain, a central nucleotide-binding domain (NACHT), and a C-terminal ligand-binding region that is composed of several leucine-rich repeats (LRRs). The NLRP and NLRC subgroups differ in their N-termini with a pyrin domain (PYD) and a caspase recruitment domain (CARD), respectively.
NLRP3 comprises a PYD-NACHT-LRR module, where the NACHT and LRR domains interact to maintain NLRP3 in an auto-repressed/closed conformation. PYD drives the inflammasome formation upon NLRP3 activation (9).
NLRP1 architecture contrasts with NLRP3 and other NLRP proteins. In addition to a PYD-NACHT-LRR module in N-terminal , it features a function-to-find-domain (FIIND) upstream of a CARD domain in C-terminal. FIIND undergoes constitutive auto-proteolysis, generating a "licensed" full-length NLRP1 (NLRP1FL), in which the N-terminal (NLRP1NT) and C-terminal (NLRP1CT) fragments remain non-covalently associated (10). Structural studies have revealed a ternary complex where one NLRP1FL and one NLRP1CT bind to dipeptidyl peptidases, DPP8/9, keeping the inflammasome-forming NLRP1CT in check (11). How the single NLRP1CT is initially generated is unknown. Upon NLRP1CT release, CARD drives NLRP1 inflammasome formation. Of note, in mice, PYD is replaced by an NR100 domain.

• NLRP3 inflammasome formation
NLRP3 can sense a wide range of stimuli (microbial, sterile, or environmental). Yet, no NLRP3 ligand per se has been described and the exact mechanisms underlying NLRP3 activation remain to be fully elucidated. As of today, the paradigm is that cytosolic stress signals such as K+ efflux and oxidized mitochondrial DNA contribute to NLRP3 post-translational modifications and opening (12,13). This change in NLRP3 conformation allows PYD to recruit pro-caspase-1 via ASC, a PYD/CARD-containing adaptor (12). The NimA-related protein kinase 7 (NEK7) interacts with LRR and has been proposed to support NLRP3 oligomerization in mice (14,15), while it is dispensable in humans (16). Importantly, NLRP3 activation steps seem highly conserved in humans and mice, allowing the study of NLRP3 relevance using murine disease models.
• NLRP1 inflammasome formation
A major gap in the comprehension of NLRP1 inflammasome was filled in 2012 when the anthrax lethal factor (LF) was shown to activate NLRP1 and drive pro-inflammatory responses upon B. anthracis infection in a mouse model (17). However, this finding did not recapitulate in humans (10). Since 2020, our understanding of human NLRP1 activation has accelerated. Most reports are in line with a "functional degradation" model: distinct stimuli, listed below, trigger destabilization and proteasomal degradation of the NLRP1NT portion, allowing the release of the NLRP1CT fragment.
– Val-boroPro (VbP, Talabostat), a small molecule known to stimulate anti-cancer immune responses, inhibits DPP8/9’s catalytic activity and induces NLRP1NT degradation both in mice and humans (11,18). Of note, there is currently no (patho)physiogical-encoded molecule identified to act as a DPP8/9 inhibitor.
– Pathogen-encoded proteins, notably proteases such as 3Cpro from the human rhinovirus, the causative agent of the common cold (3), and NSP5 from SARS-CoV-2, the causative agent of COVID-19 (4), have been shown to induce NLRP1NT degradation and NLRP1 activation. Hence, NLRP1 has been suggested to act as a broad decoy receptor of microbial virulence factors. These triggers are restricted either to human or mouse NLRP1, illustrating host and pathogen co-evolution.
– Ultraviolet B (UVB) radiation causes a ribotoxic stress response mediated by the MAP3 kinase ZAKα. This kinase initiates a hyperphosphorylation in NLRP1NT, which possibly accelerates its degradation (5).
– Long double-stranded (ds)RNA, generated upon replication of the Semiliki Forest Virus triggers human, but not mouse, NLRP1 activation (19). In this case, NLRP1 may switch conformation, allowing NLRP1NT degradation( 19). More research is still needed to identify the mechanisms by which the NLRP1CT fragment is released.

• NLRP3 and NLRP1 safeguards
Sensor activation and inflammasome formation lead to rapid and powerful inflammatory responses, extremely harmful if accidentally triggered. Thus, to prevent unwanted activation, multiple safeguards exist at all steps of the inflammasome response. Such checkpoints have been described at the sensor level, at least for NLRP3 and NLRP1.
NLRP3 activation is governed by two signals. A first priming signal renders the NLRP3 sensor responsive to a second activatory signal. The priming signal can be delivered by PRRs (pathogen recognition receptors) or cytokine receptors. Depending on the cell type, as well as the signal nature and duration, NLRP3 priming occurs through transcriptional upregulation, post-translational modifications, or binding with a protein partner, such as NEK7 (13,20). Recently, Schmacke et al. have identified an IKKβ-mediated NEK7-independent and predominant priming pathway for NLRP3 in humans (16). The authors show that IKKβ induces NLRP3 priming by increasing its recruitment to the trans-Golgi network (TGN), an organelle thought to play a key role in NLRP3 inflammasome assembly (12,16,21).
NLRP1 activation is governed by the release of the functional NLRP1CT fragment from the ternary complex DPP8/9-NLRP1FL-NLRP1CT, which has been suggested as a checkpoint for NLRP1 inflammasome formation (11,22). A recent report by Ball et al. indicates that during homeostasis, NLRP1FL is associated with oxidized thioredoxin-1 (TRX1) and that reductive stress triggers their dissociation and the proteasomal degradation of the NLRP1NT portion. The resulting NLRP1CT fragment is sequestered and kept in check within the ternary complex until a second signal triggers its release for inflammasome assembly (22). These in vitro data strongly suggest that reductive stress potentiates NLRP1 activation. Yet, additional research is needed to identify physiological inducers of reductive stress. Moreover, it is unclear whether the NLRP1NT degradation upon reductive stress acts as a priming step, or if it induces the NLRP1 inflammasome formation. In both cases, it may be the accumulation of NLRP1CT after degradation of NLRP1NT by reductive stress and other stimuli (e.g. viral proteases, UVB radiation) that unlocks the ternary complex checkpoint (11,22).
The frontier between the priming and activation for NLRP3 or NLRP1 sensors upstream of the inflammasome formation remains a major research axis.
• Signaling
Activated NLRP3 and NLRP1 sensors associate with ASC through their PYD and CARD domain, respectively, to recruit pro-caspase-1 (12,23). In humans, pro-caspase-1 interaction with NLRP1 is strictly ASC-dependent, while it is dispensable in mice (23). Ultimately, this complex causes caspase-1 activation, which in turn, cleaves pro-IL-1β and pro-IL-18, as well as Gasdermin D (GSDMD) into their biologically active forms. The formation of GSDMD pores at the plasma membrane allows the secretion of IL-1β and IL-18, and eventually leads to pyroptotic cell death (24).
Of note, NLRP3 and NLRP1 inflammasomes may use additional signaling mechanisms. In mice harboring an inactive/knockout caspase-1 or GSDMD, the NLRP3 inflammasome engages a delayed, alternative cell death, mediated by caspase-8, caspase-3, and Gasdermin E (GSDME) (25,26). In human lung epithelial cells, the SARS-CoV-2 protease NSP5 counteracts NLRP1 inflammasome signaling by inactivating GSDMD. The infected cell death is ensured by NLRP1-driven activation of caspase-3 and GSDME cleavage (4). Interestingly, patients with severe COVID-19 display elevated plasmatic levels of inflammasome markers (IL-18, GSDMD, GSDME, caspase-3) and mature IL-16, a caspase-3-derived alarmin (4). This finding warrants future in vivo studies to decipher the role and mechanisms of action for IL-16, for which little is known. Moreover, whether the caspase-3-GSDME axis is a compensatory or parallel signaling pathway for the cytokine release and/or the lytic phase, remains to be elucidated for each inflammasome (26).
Future challenges
Inflammasomes are beneficial in clearing pathogens, but their excessive stimulation can be detrimental to the host (2,27,28). Thus, it is crucial to develop fine therapeutic strategies, avoiding systemic blockade of inflammatory cytokines and the resulting opportunistic infections. Severe COVID-19 may illustrate exacerbated NLRP3 responses. Reports show NLRP3 co-staining with inflammasome specks in patient monocytes(29). NLRP3 implication in lung pathology has also been demonstrated using a mouse model of SARS-CoV-2 infection30. However, the recent finding of NLRP1 activation by the SARS-CoV-2 NSP5 protease is calling for a cautious experimental design. Is there a possibility that the data implying a direct NLRP3 response to the virus may also be attributed to the cellular stress caused by the NLRP1 inflammasome response? Likewise, NLRP3 activation may be triggered by the non-canonical caspase-4/5 response to LPS from bacterial secondary infection, a common event in respiratory viral infections (31).
Deciphering the relative importance of each inflammasome and feedforward loops during the course of infections and pathogenicities requires in vivo studies. To circumvent the poor NLRP1 conservation between mice and humans, one strategy would be to use humanized mice where murine inflammasome elements are replaced by the transgenic expression of their human counterparts.
In the absence of infections or sterile inflammation, gain-of-function (GOF) mutations in NLRP3 and NLRP1 cause monogenic human inflammatory diseases (i.e. inflammasomopathies) (32,33). Hence, such mutations in NLRP3 cause cryopyrin-associated periodic syndromes (CAPS), while GOF mutations in NLRP1 cause conditions such as NLRP1-associated autoinflammation with arthritis (NAIAD) and multiple self-healing palmoplantar carcinoma (MSHPC) (32,33).
Interestingly, most NLRP3 and NLRP1 autoinflammatory mutations lie in domains that are crucial for sensor inhibition (32,33). Therefore, they provide a first testing ground to screen small molecules for preventing inflammasome hyperactivation, by targeting specific sensors. Murine inflammatory disease models allowed the screening and discovery of MCC950, a potent and specific NLRP3 inhibitor (34,35). This promising drug has been tested in phase II in clinical trials and is awaiting for further tests. Still, it has inspired pharmaceutical companies which now have advanced clinical trials of several other NLRP3 inhibitors (36).
Many questions remain open and more studies are needed to relieve the burden of inflammasome-linked diseases on public health.
References:

1.Martinon, F. and J. Tschopp, Cell Death Differ, 2007. 14(1): p. 10-22.
2.Mangan, M.S.J., et al., Nature Reviews Drug Discovery, 2018. 17: p. 588.
3.Robinson, K.S., et al., Science, 2020: p. eaay2002.
4.Planès, R., et al., Molecular Cell, 2022. 82(13): p. 2385-2400.e9.
5.Robinson, K.S., et al., Science, 2022. 377(6603): p. 328-335.
6.Zhong, F.L., et al., Cell, 2016. 167(1): p. 187-202.e17.
7.Fenini, G., et al., International Journal of Molecular Sciences, 2020. 21(13): p. 4788.
8.Bauernfried, S. and V. Hornung, J Exp Med, 2022. 219(1).
9.Tschopp, J. and K. Schroder, Nature Reviews Immunology, 2010. 10(3): p. 210-215.
10.Chavarría-Smith, J., et al., PLOS Pathogens, 2016. 12(12): p. e1006052.
11.Hollingsworth, L.R., et al., Nature, 2021. 592(7856): p. 778-783.
12.Swanson, K.V., M. Deng, et al., Nature Reviews Immunology, 2019. 19(8): p. 477-489.
13.Groslambert, M. and B.F. Py, J Inflamm Res, 2018. 11: p. 359-374.
14.He, Y., et al., Nature, 2016. 530: p. 354.
15.Sharif, H., et al., Nature, 2019. 570(7761): p. 338-343.
16.Schmacke, N.A., et al., Immunity, 2022. 55(12):2271-2284.e7
17.Kovarova, M., et al., The Journal of Immunology, 2012. 189(4): p. 2006-2016.
18.Chui, A.J., et al., Science, 2019. 364(6435): p. 82-85.
19.Bauernfried, S., et al., Science, 2020: p. eabd0811.
20.Christgen, S., D.E. Place, et al., Cell Research, 2020.
21.Hamilton, C. and P. Anand, F1000Research, 2019. 8(676).
22.Ball, D.P., et al., Sci Immunol, 2022. 7(77): p. eabm7200.
23.Ball, D.P., et al., Life Sci Alliance, 2020. 3(3).
24.Kovacs, S.B. and E.A. Miao, Trends in Cell Biology, 2017. 27(9): p. 673-684.
25.Schneider, K.S., et al., Cell Rep, 2017. 21(13): p. 3846-3859.
26.Zhou, B. and D.W. Abbott, Cell Rep, 2021. 35(2): p. 108998.
27.Platnich, J. and D. Muruve, Archives of Biochemistry and Biophysics, 2019. 670.
28.Xue, Y., et al., Trends in Immunology, 2019. 40(11): p. 1035-1052.
29.Rodrigues, T.S., et al., Journal of Experimental Medicine, 2021. 218(3): p. e20201707.
30.Zeng, J., et al., EBioMedicine, 2022. 75: p. 103803.
31.Vora, S.M., J. Lieberman, et al., Nat Rev Immunol, 2021. 21(11): p. 694-703.
32.Alehashemi, S. and R. Goldbach-Mansky, Front Immunol, 2020. 11: p. 1840.
33.Lin, B. and R. Goldbach-Mansky, J Allergy Clin Immunol, 2022. 149(3): p. 819-832.
34.Coll, R.C., et al., Nat Med, 2015. 21(3): p. 248-55.
35.Coll, R.C., et al., Nature Chemical Biology, 2019. 15(6): p. 556-559.
36.Chen, Q.-L., et al., Biomedicine & Pharmacotherapy, 2021. 138: p. 111442.